Understanding Snakemake workflows
To begin with understanding this setup, it is highly advised to go through the basic Snakemake’s tutorial.
To surmise the workflow setup, it’s easier to follow the bottom-top approach i.e. identify the final files (or a set of) that’s required and build the way up to the infput files. This is an example of the DAG created by Snakemake.
A basic pipeline
Let’s start to setup the pipeline to run a basic set of softwares required for preprocessing a scRNAseq data i.e. use STARsolo to align our cDNA fastqs and run a set of PICARD tools while retaining all the statistics produced by these tools.
For our example case, we require 2 sets of files from our workflow i.e the outputs created by PICARD programs - CollectGcBiasMetrics and CollectRnaSeqMetrics as they are created independent of each other. Hence, if we were to use only the outputs of one program the other outputs won’t be produced.
To finally assimilate all these statistics (alignment statistics and read statistics) we will be runnning another script run_update_logs.sh (click here to know how)
Setting up
Firstly, create a list of inputs (check here for different styles of inputs) - we will go with creating text file with the list of fastq files (one line per sample).
How fastq files are arranged
This following pic shows how the fastq files are present in our directory.
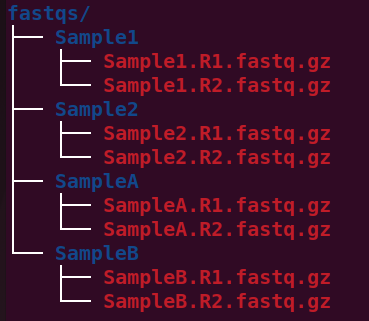
directories
Create fastq_files.txt
This following pic shows the content of the fastq_files.txt.
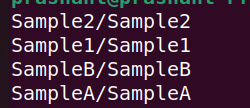
pools
As one can see it contains one representation for each sample i.e. doesn’t separate R1 and R2.
This shows how all the files are present in our directory.
tree_struct